The new version of UTRdb hosts sequences and annotations from more than 570 organisms for a total of 26,098,657 annotated UTRs.
All the UTRdb entries have been generated according to the sequence data and gene annotations available on Ensembl databases.
Searching into UTRdb is quite straightforward and also users with no bioinformatics skills can perform accurate searches across the database by:
UTRs annotations are stored according to their genomic context and can be retrieved by providing the UTR_type (5'/3' UTR, 5' UTR, 3' UTR):
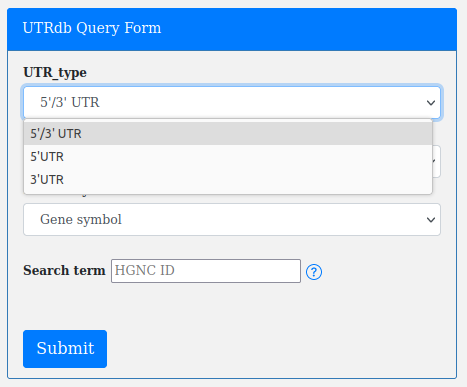
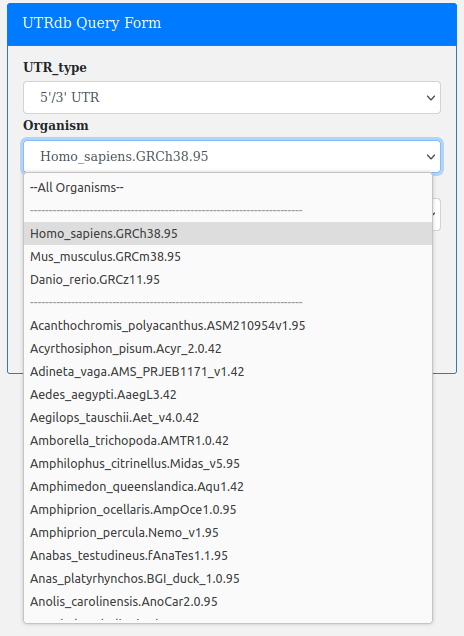
The Organism:
Note. Select "All Organisms" if not interested in a particular species.
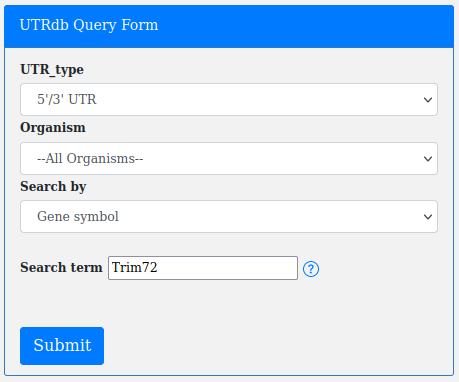
A genomic locus (Search by Gene symbol):
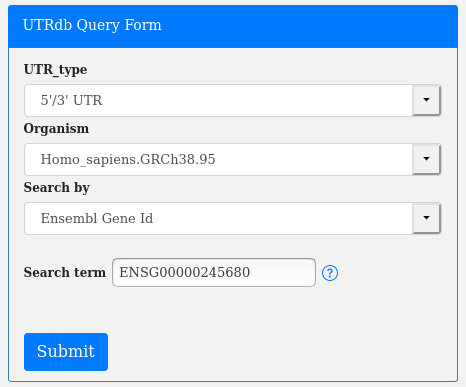
Or an Ensembl Gene Id (Search by Ensembl Gene Id):
Or an Ensembl Transcript Id (Search by Transcript Id):
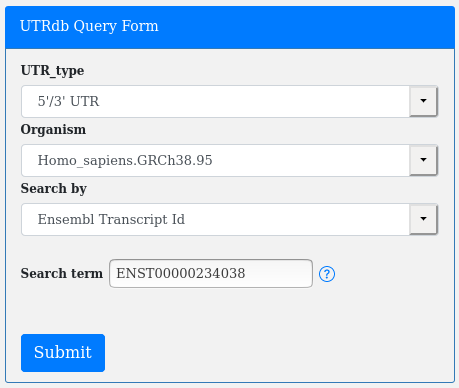
A simple helper is available on the right of the search term box:

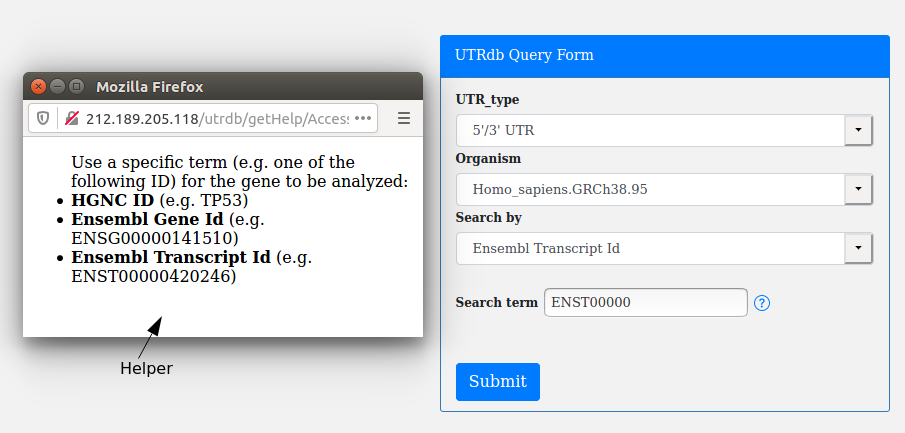
Search by Gene symbol, Search by Ensembl Gene Id activate the "autocomplete function".
This in order to facilitate the selection of the right gene, Ensembl Gene Id or Ensembl Transcript Id.
Note. The autocomplete function is available for each of the organisms stored in UTRdb. Since it changes according to the genetic
makeup of each organism, it doesn't work with the 'All organisms' selection.

The basic functions provided by the Simple retrieval form can be extended by using the filters included with the Advanced retrieval form.
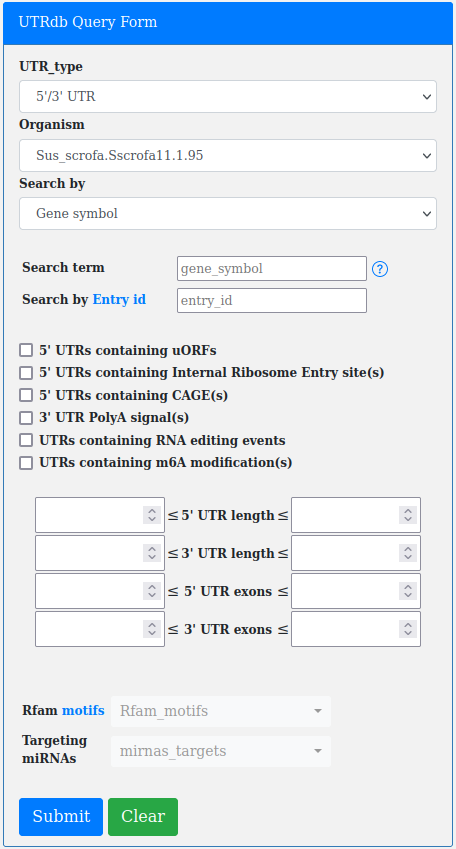
The advanced query form incorporates the functionality of the simple one and extends it.
In particular, since all UTRdb records are cataloged through a unique identifier, whose main structure is:
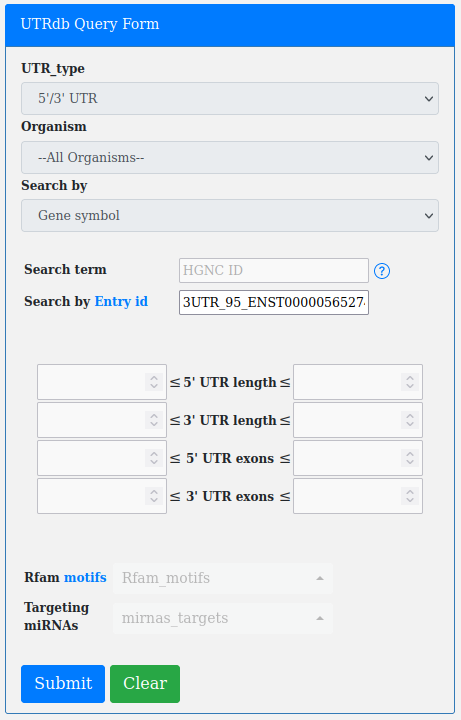
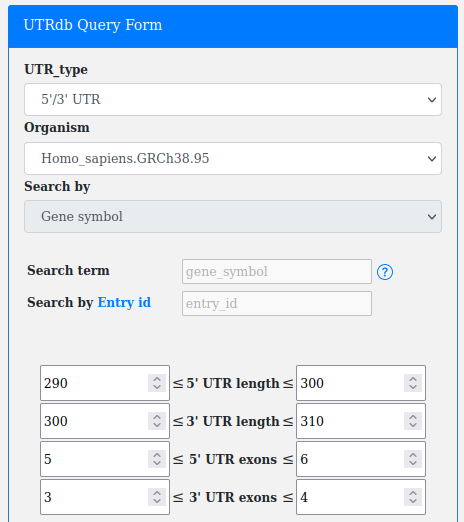
An important filter available through the advanced query form is the "Search by UTR length".
The user can search for UTRs according to a minimum and a maximum lenght (both expressed in base pairs).
The length in each box can be set manually by copy/paste and adjusted by the arrows ▲ ▼ present in each box.
The box on the left is the minimum length, while the box on the right is the maximum.
The number of boxes (2 boxes/4 boxes) changes according to the UTR_type selection (5'/3' UTR, 5' UTR, 3' UTR).
Note. The "Search by UTR length" coupled with "All Organisms" may take time (especially with very wide ranges of lengths.)

An additional filter present in the advanced query form is the "Search by UTR exons number".
This filter is very similar in operation to the search by length.
The user can search for UTRs according to a minimum and a maximum number of exons.
The numbers in each box can be set manually by copy/paste and adjusted by the arrows ▲ ▼ present in each box.
The box on the left is the minimum, while the box on the right is the maximum.
The number of boxes (2 boxes/4 boxes) changes according to the UTR_type selection (5'/3' UTR, 5' UTR, 3' UTR).
Note. The "Search by UTR exons" can be combined with the "Search by UTR length".
This combination, coupled with "All Organisms" may take time (especially with very wide ranges of lengths and exons numbers)
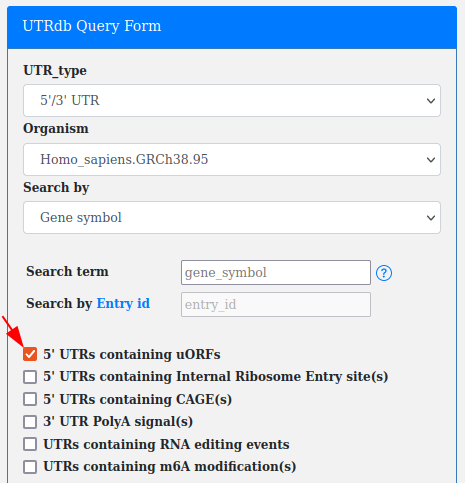
Upstream open reading frames (uORFs) are open reading frames (ORFs) within the 5'UTRs.
A checkbox in the advanced query allows for searching all the UTRs containing annotated uORFs.
This filter can be combined with "UTR_type", "Organism" and "Search by Gene_name/Ensembl Gene Id/Ensembl Transcript Id".
According to uORFs biological localization the uORF checkbox is available only with 5'/3' UTR and 5' UTR selection.
Note. This filter can be used as is or in combination with others (e.g. RNA editing, IREs...).
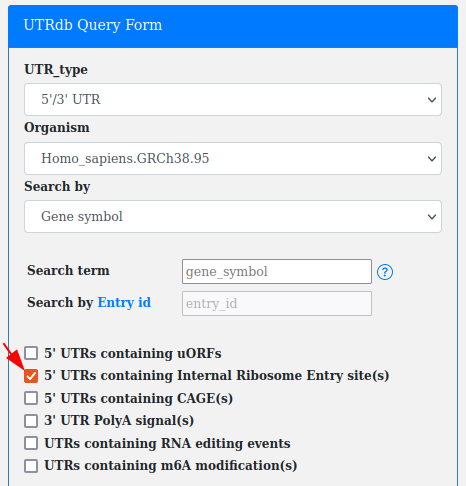
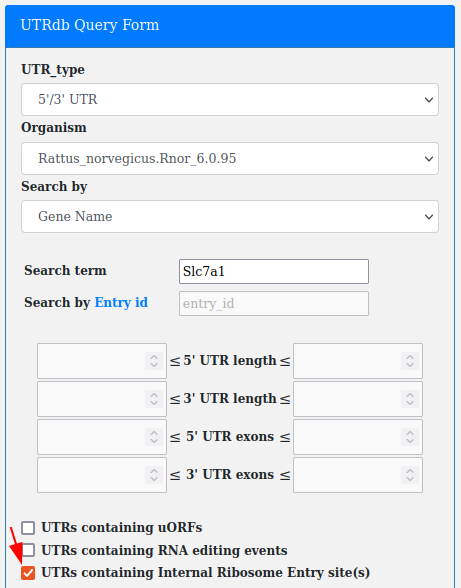
Internal ribosome entry sites (IREs), are RNA elements allowing for translation initiation.
They tipically occur in the 5'UTRs.
A checkbox in the advanced query allows for searching all the UTRs containing annotated IREs (from IRESite).
This filter can be combined with "UTR_type", "Organism" and "Search by Gene_name/Ensembl Gene Id/Ensembl Transcript Id".
According to IREs biological localization the IREs checkbox is available only with 5'/3' UTR and 5' UTR selection.
Note. This filter can be used as is or in combination with others (e.g. RNA editing, IREs, CAGEs...).
CAGE (Cap Analysis of Gene Expression) is based on a series of full-length cDNA technologies previously developed at RIKEN.
The purpose of the technology is to comprehensively map the vast majority of human transcription starting sites and hence their promoters.
A checkbox in the advanced query allows for searching all the UTRs containing annotated CAGEs (from Fantom).
This filter can be combined with "UTR_type", "Organism" and "Search by Gene_name/Ensembl Gene Id/Ensembl Transcript Id".
According to CAGEs biological localization the CAGEs checkbox is available only with 5'/3' UTR and 5' UTR selection.
Note. This filter can be used as is or in combination with others (e.g. RNA editing, IREs, m6a...).

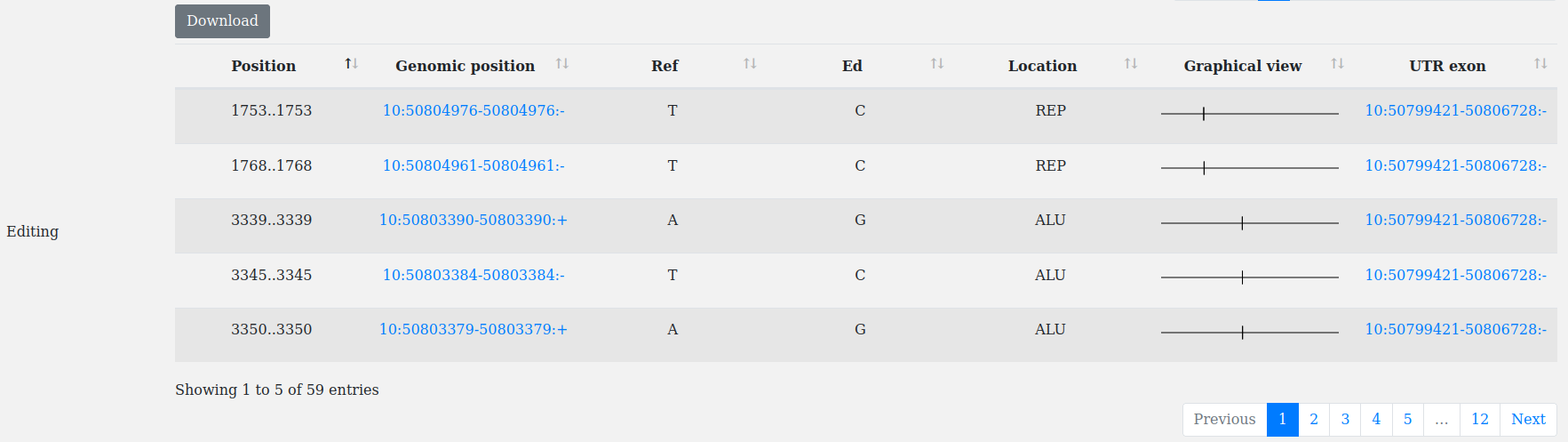
RNA editing in untranslated regions (UTRs) regulates mRNA stability/expression.
A checkbox in the advanced query allows for searching all the UTRs containing annotated RNA editing events (A-to-I deamination from REDIportal).
This filter can be combined with "UTR_type", "Organism" and "Search by Gene_name/Ensembl Gene Id/Ensembl Transcript Id".
Note. This filter can be used as is or in combination with others (uORFs, IREs...).
Rfam is a database containing information about non-coding RNA (ncRNA) families and other structured RNA elements.
Rfam derived annotations, falling whithin 5'/3'UTRs can be retrieved by using the Rfam motifs dropdown menu.
This filter works in combination with "UTR_type" and "Organism".
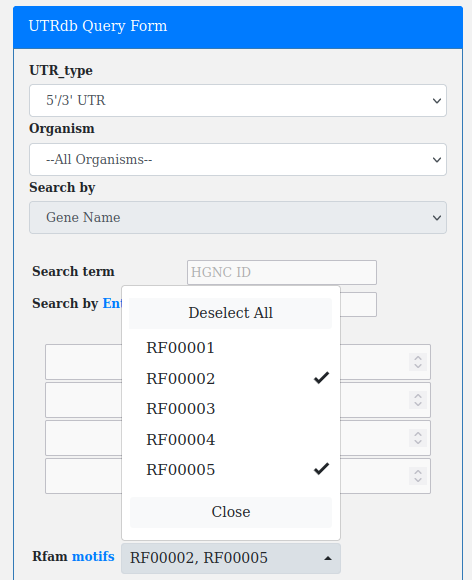
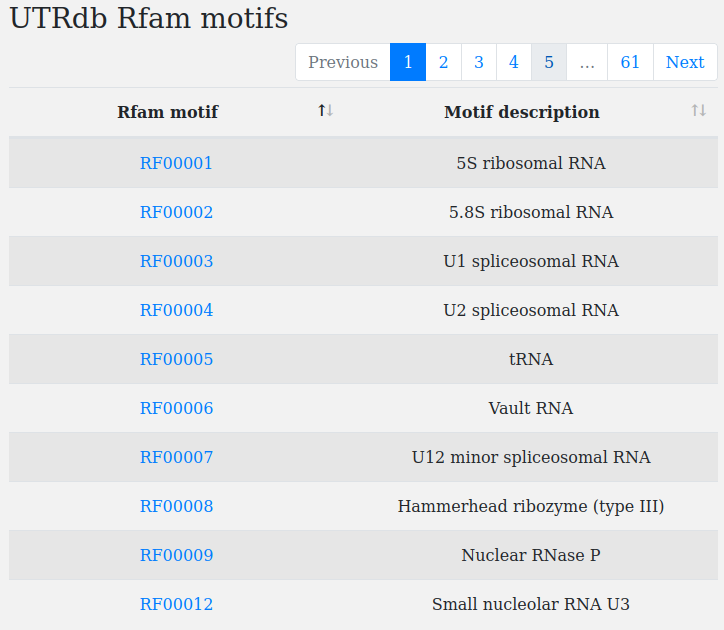
Note. For a complete list of all RFAM motifs and their description click on "motifs".
The click redirects to a page containing a table with rfam motifs IDs, motifs short description and a link (e.g click on RFAM00001, RFAM00002, etc.) to their Rfam respective pages.
MicroRNAs (miRNAs) tipically bind to the 3'UTRs of their target mRNAs interfering with translation.
UTRs stored in UTRdb, whose coordinates (according to TarBase) are experimentally recognized as miRNA targets, can be easily retrieved
by using the Targeting miRNAs dropdown menu.
This filter works in combination with "UTR_type".

The advanced query form can be reset to the default parameters (5'/3'UTR, All organisms) by clicking on the clear button.

UTRs stored in UTRdb can be explored in their genic context through a custom "Gene View module" based on Python3, Javascript HighCharts library and HTML5.
The "Gene View" page can be accessed through the main Website Menu.

The Gene View page contains a small form that allows searches by Organism, Gene symbol, Ensembl gene id, Ensembl Transcript id.
As with all other UTRdb forms (except for "All organism") search term autocompletion module is active.
Note.If "All organism" is selected, the search will be performed on the entire organism database.
After filling/selecting the required fields and clicking on submit, a Results page will be returned.
Note.If "All organism" and "Search by Gene symbol" are selected, the query will return all the entries for that gene in the different organisms
stored in UTRdb.
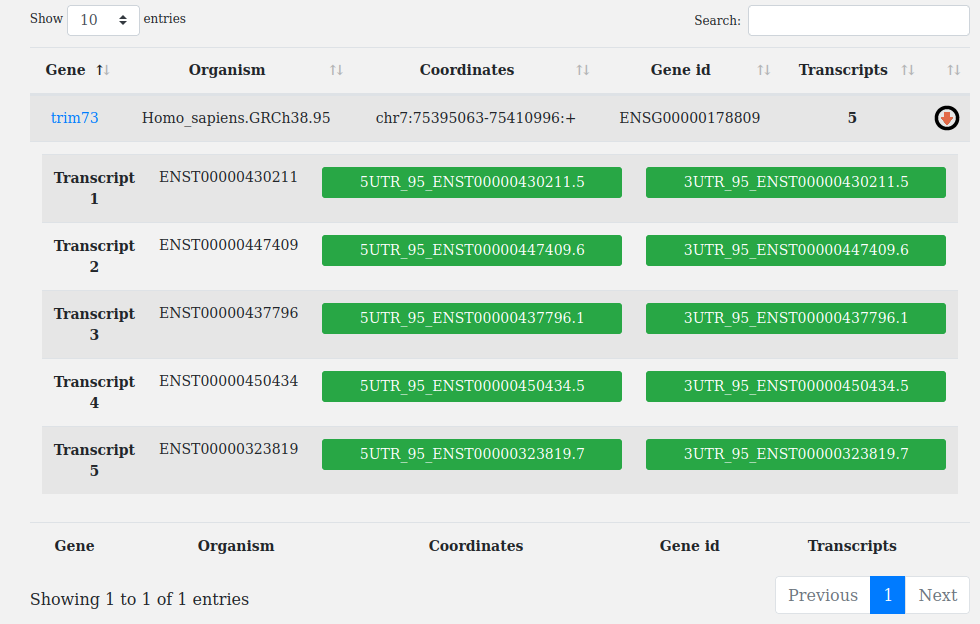
The results page is in tabular format and contains the following columns:

To show the nested pop-up table of isoforms, just click on the arrow  at the end of each row.
at the end of each row.
The nested table contains the different isoforms of the gene (with their ensembl identifiers) and the associated UTRdb entries.
If for a given transcript, UTRs are reported, the name of the UTRdb entries will be within a green rectangle, otherwise there will be a red rectangle with the words no 5'UTR or no 3'UTR.
By clicking on the UTRdb entries the user will be redirected to the corresponding UTRdb entry record. See below in the page for further details.
By clicking on the gene_name (e.g trim73) the user will be able to see the structure of the transcripts reported in the results page.
According to colours legend, in the Transcripts Diagram:
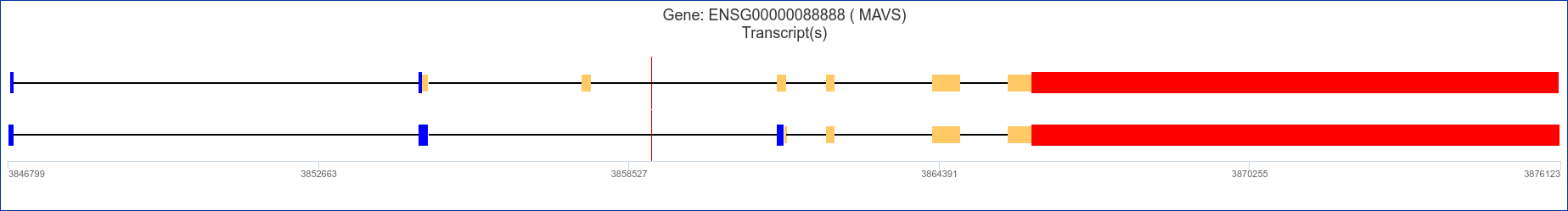
By mousing over each transcript diagram block, the corresponding information will be displayed.
By clicking on the UTRs blocks the user will be redirected to the corresponding UTRdb entry record. See below in the page for further details.

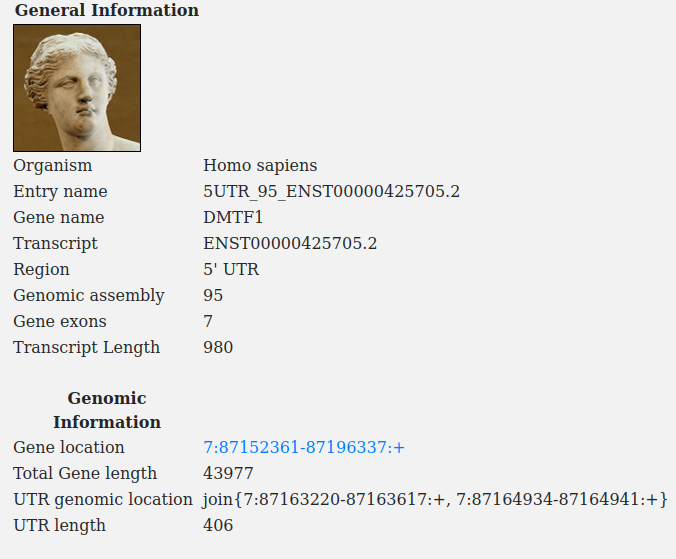
A typical utrdb entry consists of 3 main parts:

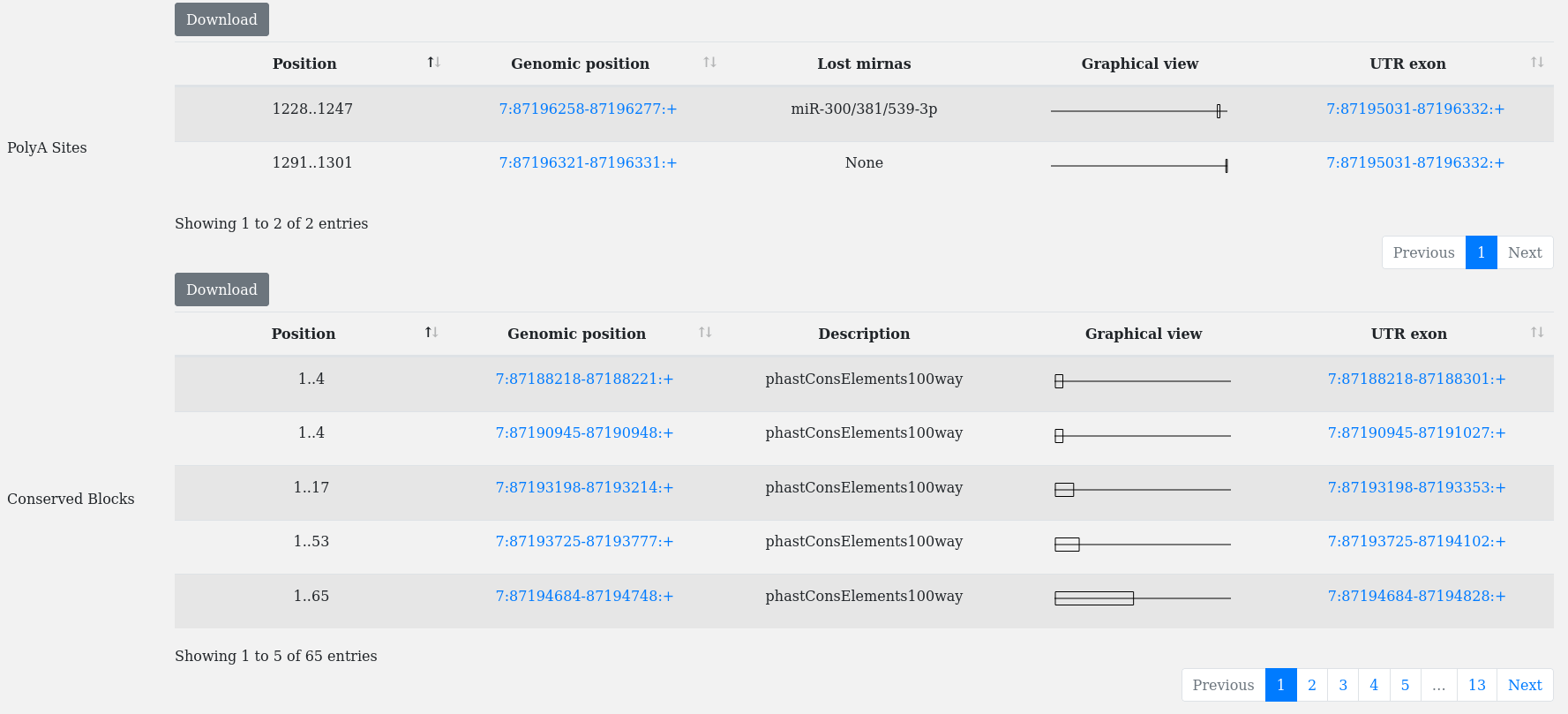




 button.
button.
